随着中国药典和GMP要求的不断发展完善,无菌检查隔离器日益受到国内制药企业的关注。本文介绍了无菌检查隔离器的相关法规要求、发展技术背景及设备特点等。
1、无菌检查隔离器的法规与技术背景
1.1与无菌检查隔离器相关的法规
中国药典2010年版附录XIH无菌检查法中规定,“无菌检查应在环境洁净度10000级下的局部洁净度100级的单向流空气区域内或隔离系统中进行”。而2014年中国药典对微生物实验室的增补征求意见《附录XVIG药品微生物实验室质量管理指导原则》中说明,“无菌检查应在B级背景下的A级单向流洁净区域或D级背景下的隔离器中进行”。但中国药典2015年版通则微生物内容第3次公开征求意见的通知中《1101无菌检查法》中取消了第2次公开征求意见中对环境要求的明确说明。值得注意的是新增加了《9206无菌检查用隔离系统验证指导原则》,这是中国药典中首次纳入隔离器,其中提到“无菌检查用隔离器安装环境的洁净度要求建议不低于我国现行GMP中D级空气洁净度要求,安装隔离器的房间应限制无关人员出入”。由此可见,隔离器在无菌检查中的应用己成为趋势。
以下是国际主流法规机构对无菌检查的相关说明。
(1)美国药典(USP)24版无菌检查-隔离器系统验证:“用于实施药典所规定无菌检査的隔离器从1980年代中期就开始使用了。……使用隔离器的操作者无需穿着特殊的洁净服来操作无菌检查,标准的实验室服就足够了。……无菌检査隔离器不一定要放置于定级的洁净室中,重要的是放置隔离器的区域要对非必要人员的进出限制管理。……房间里无需进行环境监控。……”
(2)国际药品认证合作组织(PIC/S)PI012-3无菌检查:
“无菌检查应该在无菌条件下进行,无菌检查的操作环境要求和无菌药品的生产环境一致。”
(3)PIC/SPI014-3用于无菌工艺生产和无菌检查的隔离器:
“当隔离器用于无菌检查时,没有正式要求他们被放置在D级环境。这个环境应当受到控制,例如只允许受过培训的员工进入,但不一定需要定级别。”
1.2无菌检查隔离器的发展与优势
在隔离器技术用于无菌检查之前,执行无菌检查的操作和产生假阳性是关注的重点问题。无菌检查过程中,假阳性的产生有以下可能性:(1)由操作者在无意识的情况下造成的污染;(2)在容器表面存在的污染;(3)测试环境与设施造成的污染;(4)使用受到污染的试剂和设备,或者取样的操作产生的污染。
质量控制微生物实验室依赖于有效控制的测试环境、适当的产品样本和测试试剂,以及操作者的无菌技巧,才能够保证无菌检查的成功执行。适当的试剂和产品样本可以通过每批次促生长以及产品样本抗菌性和抗真菌性测试来进行验证,但是控制测试的环境和管理控制操作人员的行为则相对困难。随着隔离器应用于无菌检查,虽然操作方式上并没有简化,有时甚至操作起来不如传统方式方便,但是受到更可控环境的保护,加强了微生物的控制,并且降低了假阳性的发生,从而提高了测试性能。
美国某药厂对分别在洁净室及隔离器中进行无菌检查所产生的假阳性和经济损失的比较结果显示,在洁净室中进行无菌检查造成的平均假阳性产生率、未通过审查的药品率及每年的损失分别为0.5%〜1.0%、1%〜2%和20万〜50万美元;而在隔离器中进行无菌检查上述情况均可避免。另外,从使用成本角度考虑,在国际制药工程协会(internationalsocietyforpharmaceuticalengineering,ISPE)的《无菌产品生产设施基础指南》中提到,在使用隔离器时,运行成本大约低于传统洁净室运行成本的75%,主要是与供热通风与空气调节(heating,ventilationandairconditioning,HVAC)有关的运行成本;其他成本,如更衣室、厂房设施布局及环境监测方面也节省了花费。
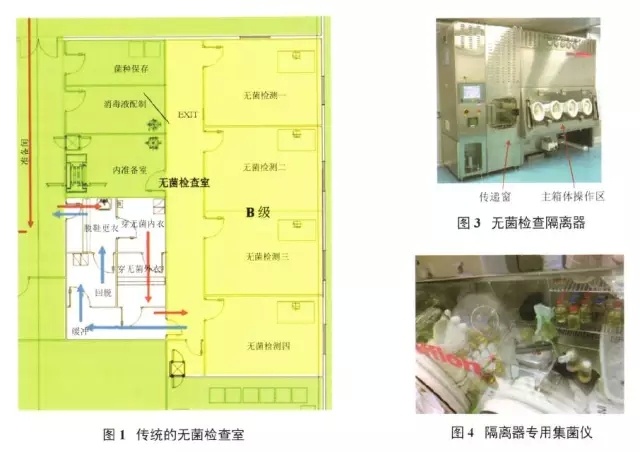
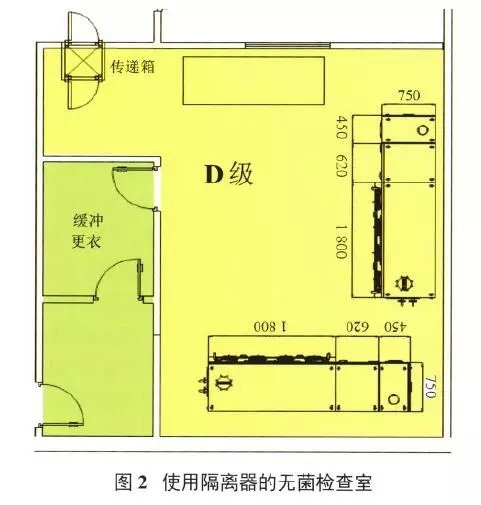
无菌检查在B级背景下的A级设计的实验室需要设计复杂的更衣区域和缓冲间(图1)。而如果使用隔离器,最简单的设计就是1个D级房间,带1个物料进出口,1个人员进出口即可(图2)。
2、无菌检查隔离器
2.1无菌检查隔离器的设计
无菌检查隔离器一般由1个传递窗和1个操作区主箱体组成,结构使用不锈钢制成(图3)。隔离器具有良好的密闭性,运行时内部正压控制,内部单向流设计。主箱体上配备有手套,用以在隔离器中执行无菌检查操作。主箱体内部设置有无菌检查专用的集菌仪,应当选用针对隔离器专门设计的集菌仪,如Millipore的Steritest™EquinoxIsofitPump(图4)。传递窗和主箱体配备内置式汽化过氧化氢灭菌系统,根据工艺需求对隔离器内部或传递进入的耗材、样品和试剂容器表面进行灭菌。
隔离器可集成粒子计数器、微生物采样器等环境监测设备,通过触摸屏进行操作,根据客户不同需求提供灭菌数据和环境监测数据报表的存储、电子版本输出或打印等功能。控制系统的设计可满足良好自动化生产实践指南(goodautomatedmanufacturingpractice,GAMP),以及21CFRPart11(美国FDA联邦法规21章第11款)对电子记录和电子签名的要求。
2.2汽化过氧化氢灭菌
汽化过氧化氢灭菌是无菌检查隔离器中最重要的功能之一,可使隔离器内部控制在1个生物负载很低的环境下。合理的灭菌循环可以使整个灭菌循环快速和安全。
由内部气流循环系统将快速生成的汽化过氧化氢带入隔离器主箱体和传递窗中,对试剂样品、耗材外包装表面进行灭菌。在隔离器完成空载的灭菌性能确认后,需要针对固定的试剂、耗材等物品的排放方式进行满载的灭菌循环开发并验证,图5为正在进行满载测试。合理的物品排放、恰当的灭菌循环工艺参数能在较短的时间内(视装载量和隔离器空间大小而定,一般为1.5〜2.5h)达到生物指示剂下降6个对数的效果。连续这种灭菌过程具有重演性,隔离器在空载时1次灭菌循环曲线见图6。
汽化过氧化氢灭菌工艺中很重要的一点是通风后过氧化氢的残留,一般要求通风后过氧化氢浓度小于1mg/L。过多的残留可能会导致假阴性的产生。
2.3有关工艺耗材、试剂和工具
图7所示为传统的无菌检查,操作人员正用注射器在酒精灯的火焰保护下处理粉针剂样品。而试剂、培养基等是用牛皮纸覆盖用线扎的。这样的传统操作方式,以及选用的耗材包装在隔离器中是绝对不推荐的。
操作者应通过隔离器的手套对无菌检查进行操作,在使用前后都要对手套的完整性进行检查。戴隔离器手套进行操作会降低手的灵活性,因此在隔离器中应当不接触或小心使用尖锐物品,如西林瓶的铝盖、针头、剪刀等,避免这些尖锐物扎破手套,导致隔离器的完整性被破坏。
所有传入隔离器的物品均须使用汽化过氧化氢进行表面灭菌,要确保表面灭菌效果的同时,还要考虑过氧化氢在包装表面造成的渗透和吸附问题。包装材料的渗透可能会造成一定的工艺影响,例如用于环境检测的培养基,如果过氧化氢渗透到培养基中,可能会造成检测结果的假阴性;而材料吸附会延长过氧化氢的分解,延长灭菌后的通风时间。因此需要对选用的包装材料进行一些评估,或者选用隔离器专用的成品耗材,如MerckMilliporeBD的相关产品。